Medical Doctor IRCCS Istituto Tumori "Giovanni Paolo II"
Introduction: Accessory stromal and immune cells are implicated in multiple myeloma (MM) pathogenesis, although the predictive role of peculiar tumor microenvironment (TME) cells toward specific treatments has been not fully clarified. Recent computational tools allow to infer quantitative and qualitative features of TME starting from bulk transcriptomic data, enabling associations of peculiar cellular components with patients’ prognosis. The aim of this work was the development of a prognostic molecular model based on TME factors to improve risk stratification of MM patients.
Methods: A total of 59 real-life pre-treatment BM aspirates underwent immunomagnetic separation of the CD138-negative cells to generate GEP by microarray (Clariom S). This training cohort included patients receiving induction therapy by bortezomib, cyclophosphamide and dexamethasone (VCD; n=55) or bortezomib, melphalan, prednisone (VMP; n = 4), and showing stringent complete response/complete response (responders; n=24) or stable disease/progression disease (non-responders; n=35). Molecular information, including cytogenetic alterations (i.e., t(4;14), t(14;16), t(14;20), amp(1q), and del(17p)) and clinical stage (R- ISS) were available for each case. We applied the CIBERSORTx to the CD138-deriving GEP to digitally purify specific stromal and immune cell types as well as their transcriptional profiles. Thus, we identified differentially expressed genes (DEGs) comparing GEP of each purified TME cytotype between the responders and non-responders subgroup, and applying a fine-tuned statistical filtering to obtain a final gene signature. This signature was finally used to build a PFS-based predictor by a penalized Cox regression.
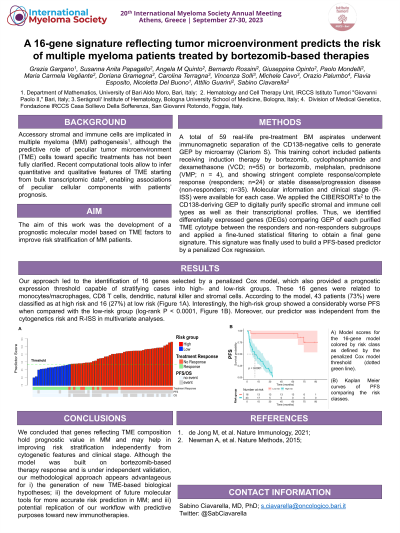
Results: Our approach led to the identification of 16 genes selected by a penalized Cox model, which also provided a prognostic expression threshold capable of stratifying cases into high- and low-risk groups. These 16 genes were related to monocytes/macrophages, CD8 T cells, dendritic, natural killer and stromal cells. According to the model, 43 patients (73%) were classified as at high risk and 16 (27%) at low risk. Interestingly, the high-risk group showed a considerably worse PFS when compared with the low-risk group (log-rank P < 0.0001). Moreover, our predictor was independent from the cytogenetics risk and R-ISS in multivariate analyses.
Conclusions: In conclusion, we demonstrated that genes reflecting TME composition hold a remarkable prognostic value in MM and may help in improving patient risk stratification independently from cytogenetic features of tumor compartment and clinical stage. Although the model was built on bortezomib-based therapy response and is under independent validation, our methodological approach appears advantageous for i) the generation of new TME-based biological hypotheses; ii) the development of future molecular tools for more accurate risk prediction in MM; and iii) potential replication of our workflow with predictive purposes toward new immunotherapies.